Preamble
This is more or less my thoughts about how to structure a bulk loader for chado.
Majority of the ideas come from writing obo2chado loader. obo2chado still lack the design that
i am aiming now, but most of the upcoming one will follow that. And the future idea is to refactor the obo loader to that mold.
Design
Scope and expectation
- The input would be some sort of flat file.
- The data will be loaded to a relational backend. It could definitely be generalized, but at this moment it is not considered.
Reading data
There should be an object oriented interface for reading data from flat files. That object is expected to be passed
along to other classes. For example, for obo2chado loader i have used the ONTO-Perl module.
Database interaction
Probably one of the import one. It’s better to have an ORM that supports mutiple backends and bulk loading support. For Perl
code, i have used BCS a DBIx::Class class layer for chado database.
Loading in the staging area
This part is supposed to get data from flat file to temp tables of RDBMS. To start with, lets assign a class which
will manage everything related to this task. First lets figure out what kind of information the class needs in order to perform those tasks.
For the sake of understaing we name it as StagingManager ….
Staging manager
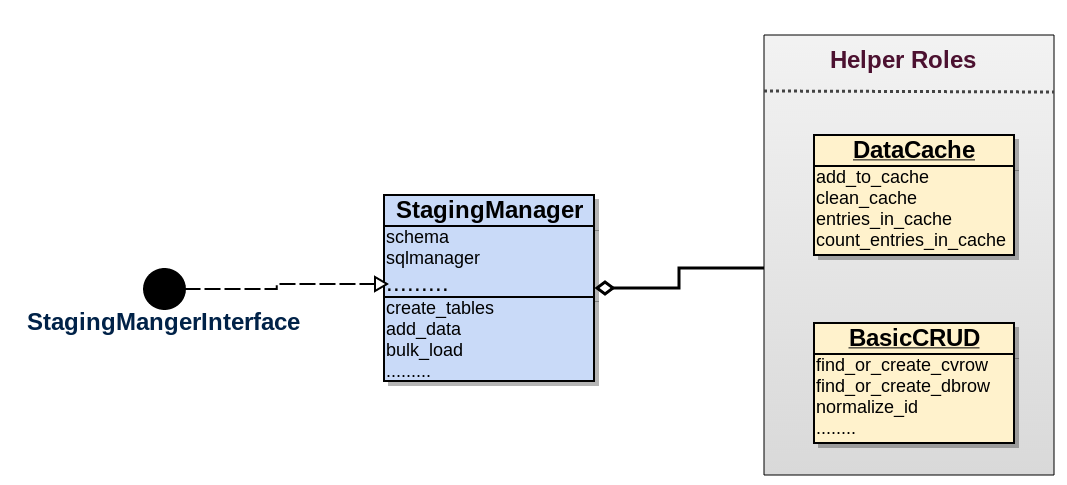
Attributes
- schema: Should have an instance of
Bio::Chado::Schema. A ORM/Database object for all database centric tasks. If its an ORM, then it should better provide access to some bulk mode operation or at least low level objects for bulk support. - chunk_threshold: I kind of thrown this in, it will be used for bulk loading in chunk.
- sqlmanager: Should have an instance of
SQL::Lib. A class that manages handling of sql statements. I found it easy to manage instead of inlining it in the class itself. With growing sql statments, it could become cumbersome to navigate through code. Provides better separation between code and non-code content. Forobo2chado, i have used SQL::Library module, seems to be a very good choice. - logger: An instance of an logger.